Kinact - Help
Help! Try using the key arrows in your keyboard or the buttons below to switch among the tips below.
Main page

About Kinact
Kinact is a novel computational method for predicting activating mutations in kinases. It is a machine learning approach that relies on structural and sequence data of proteins, such as environment residue, stability changes predictions, atomic interactions, graph-based signatures and residue conservation.
Such a predictive model is not only of great relevance for antibody engineering and development, but would also allow the prediction of biologically relevant mutations when investigating diseases, such as cancer.
Submission page
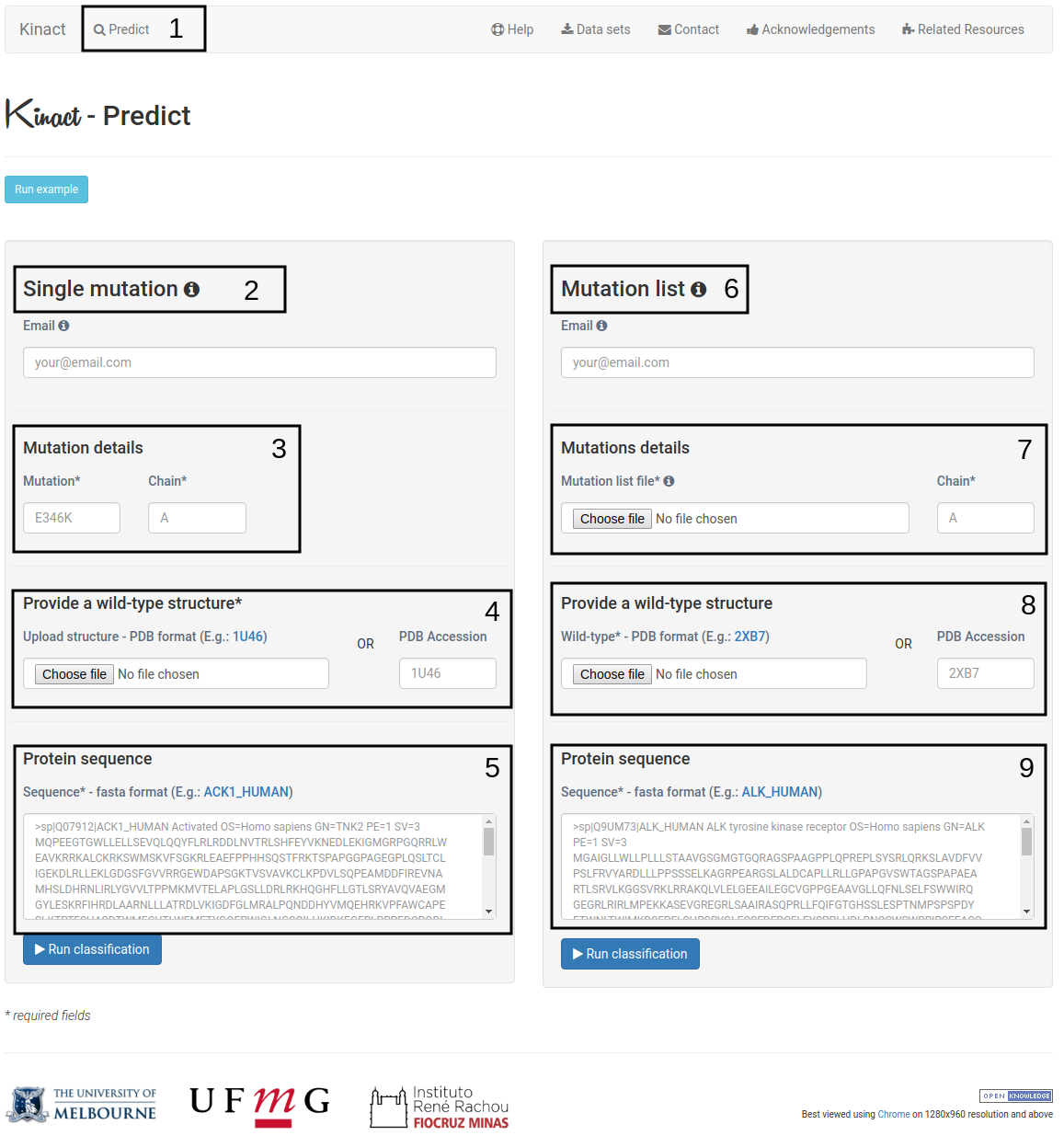
How to run a prediction
To run a prediction:
- Click on "Predict"(1) to open the submission page.
- The Single mutation option (2) allows the user to submit one single mutation for prediction.
- Details on the single mutation should be provided (3). A mutation code consists of wild-type code, residue position and mutant code (using the one letter amino acid code). Residue position must be consistent with the PDB file. The chain for the mutation code must also be provided.
- Users should also provide a wild-type structure (4) by uploading a file in PDB format or providing a PDB accession code.
- In addition, Kinact requires a protein sequence (5)that will be used for homology modelling of missing residues on the wild-type structure.
- The Mutation list option (6) allows users to submit multiple mutations to be analyzed by Kinact.
- Users must provide a list of mutations (7) by uploading a file containing the mutation codes (wild-type code, residue position and mutant code). The file format requires one mutation per line. In addition, a chain identifier, must also be provided.
- Similarly to the Single mutation option, a wild-type structure must be provided (8) by uploading a file in PDB format or providing a PDB accession code , alongside with the protein sequence (9) for homology modelling.
Results page

Results - Prediction Outcome
The results for a single mutation will be displayed once computations are completed.
- The predicted outcome by Kinact for a single mutation is showed on the left (1).
- Effects of Mutation on Stability (ΔΔG) calculated by mCSM, SDM and DUET are also shown (2) in order to provide additional information on the effects of the given mutation.
- Mutation details is displayed (3) highlighting the wild-type residue, position number and chain. Also more details regarding the protein submitted are shown (4) such as the type of secondary structure in which the wild-type residue is placed.
- (5) Provides an image representing the superposition of all 3D structures of proteins within the same group o kinases. α helix is displayed in pink, β sheets in blue and loops in gray.
- Further analysis regarding interatomic interactions of the wild-type residue, conservation and multiple sequence alignment within homologue group of kinases are also provided (6).
- Pymol Session, PDB structure and Multiple Sequence Alignment files used to generate all analysis are also available for download (7).
Results page
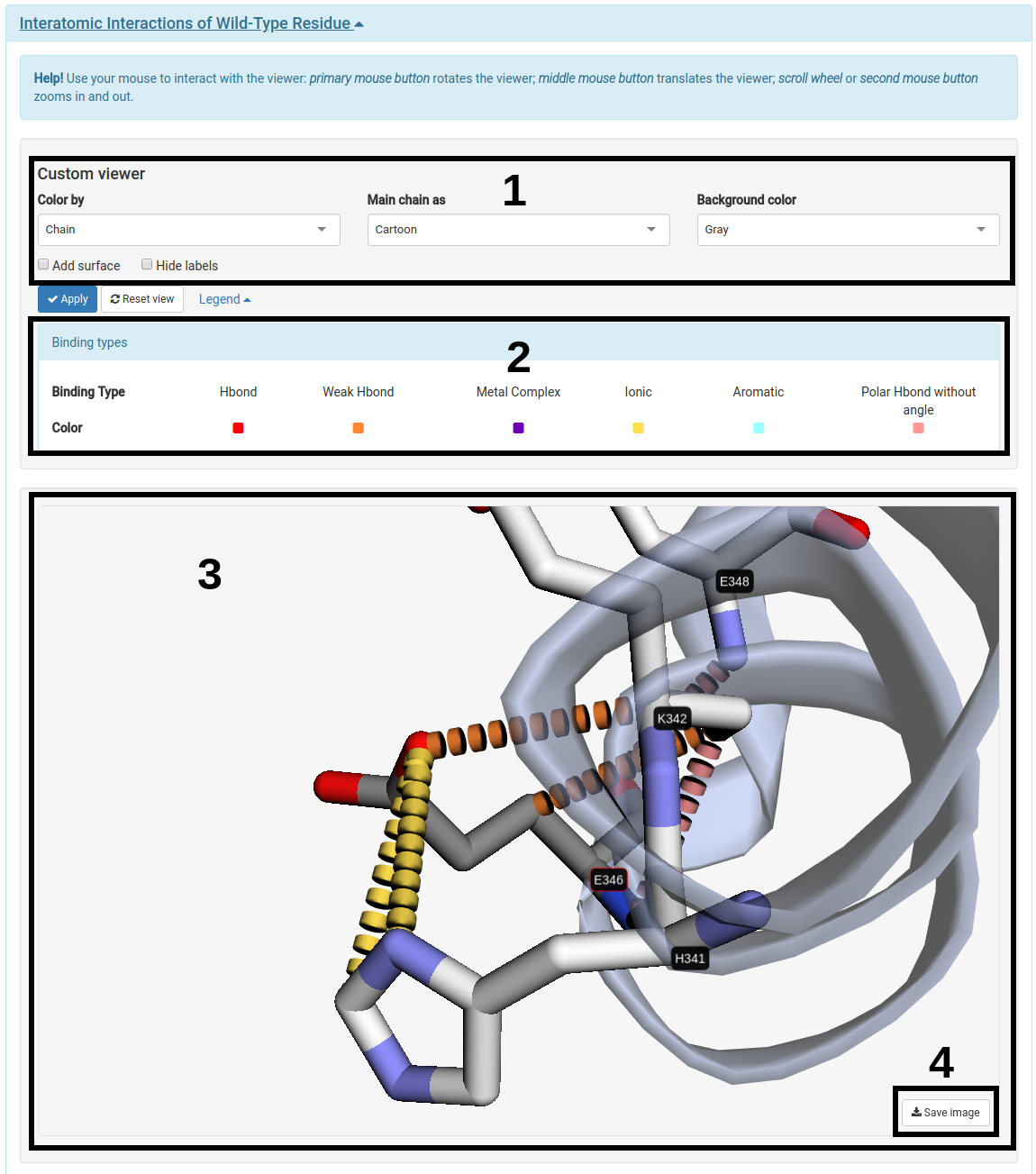
Results - Interatomic Interactions of Wild-Type Residue
For this analysis one have to click on the "Interatomic Interactions of Wild-Type Residue" bar to open the panel with the interactive 3D viewer.
- Users can customize the viewer with the options provided on the panel (1).
- The legend with the color definition for each binding type is displayed to guide the user (2).
- The viewer allows users to interact with the structure through mouse clicks and scrolling (3).
- (4) allows users to take screenshots of specific positions of the viewer.
Results page
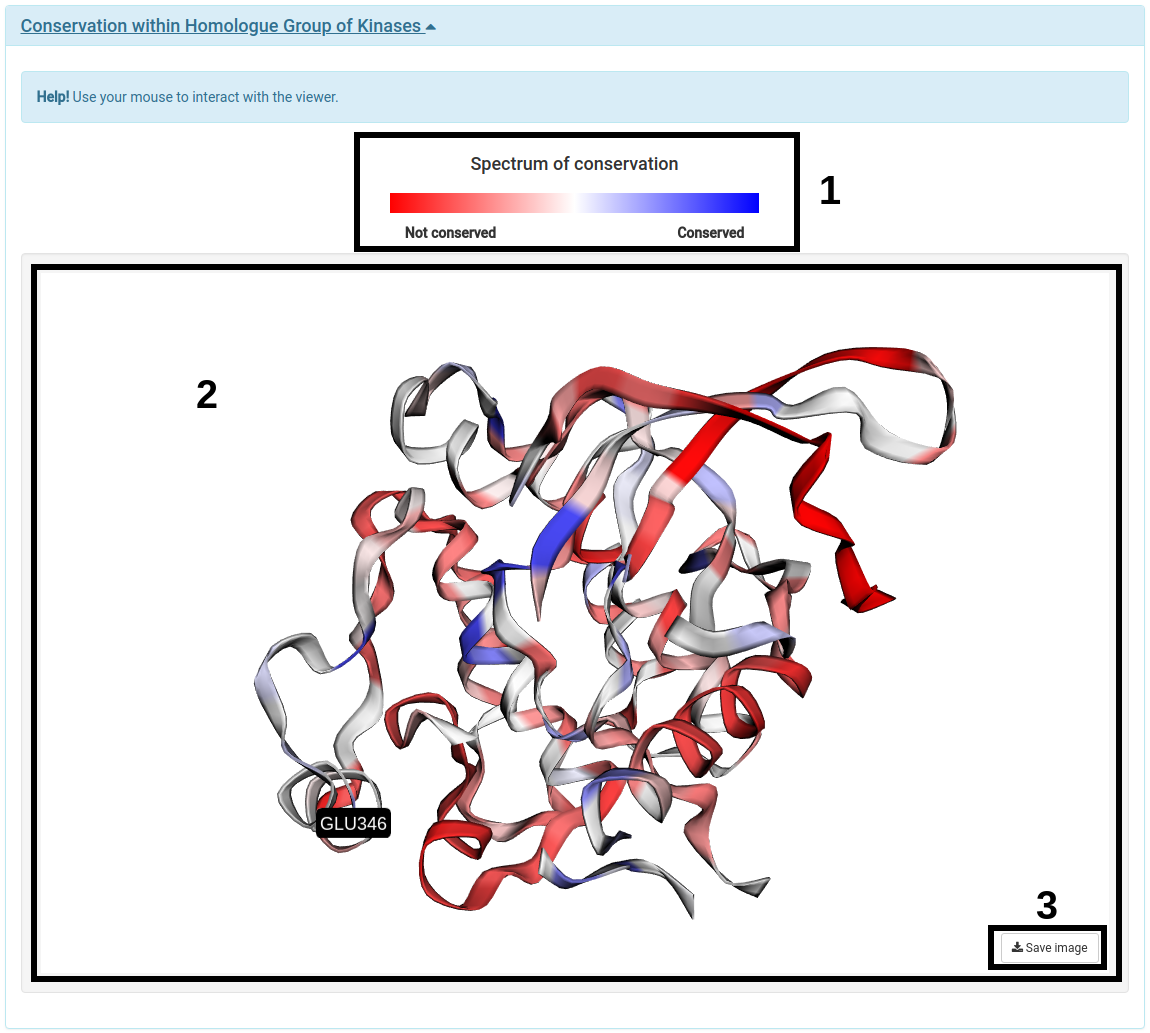
Results - Conservation within Homologue Group of Kinases
For this analysis one have to click on the "Conservation within Homologue Group of Kinases" bar to open the panel with the interactive 3D viewer.
- The legend with the color definition for the spectrum of conservation is displayed to guide the user (1).
- The interactive viewer shows the 3D structure of the chain in which the mutation specified by the user occurs is coloured according to the conservation of each residue within the Homologue Group of Kinases in which the submitted protein was classified into (2).
- (3) allows users to take screenshots of specific positions of the viewer.
Results page

Results - Multiple Sequence Alignment within Homologue Group of Kinases
For this analysis one have to click on the "Multiple Sequence Alignment within Homologue Group of Kinases" bar to open the panel with the Multiple Sequence Alignment.
- The legend with the color defition for the MSA is shown in order to guide the user (1)
- The MSA table shows the submitted protein as its first row followed by the sequences of proteins within the Homologue Group o Kinases (2).
- Only the last digit of residue numbers are shown due to visual purposes, but users can hover the cursor over any digit to see the exact number for an specific position.
- Activating mutations reported on the literature and used to build Kinact predictive model are shown in red in the MSA table. Users can hover the cursor over these to show their details.
Results page
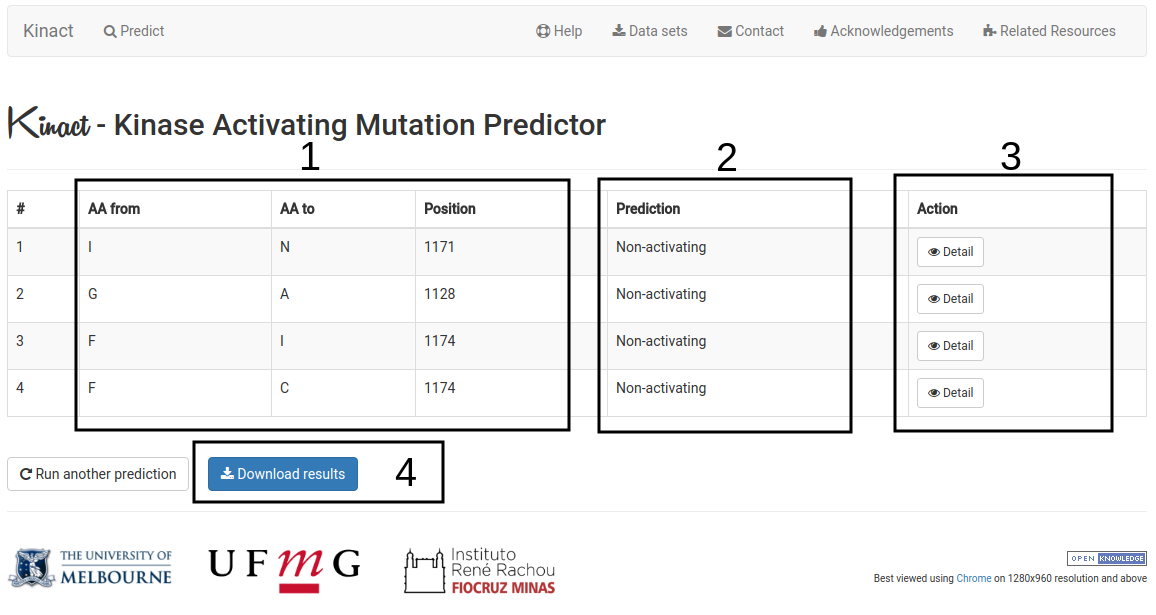
Results - List of Mutations
Your results for a list of mutations will be displayed in a table format with the following information:
- Mutation details are displayed (1).
- The predicted outcome for each mutation (2).
- Complete analyses can be performed for each one of the mutations on the list using the Detail button (3). Similar to the analysis performed for the Single mutation option.
- An option to download the predictions as a tab-separated file is also available (4).
Contact page
Getting in touch
In case you experience any trouble using Kinact or have any suggestions or comments, please do not hesitate in contacting us either via e-mail (1) or through the online form (2) .